
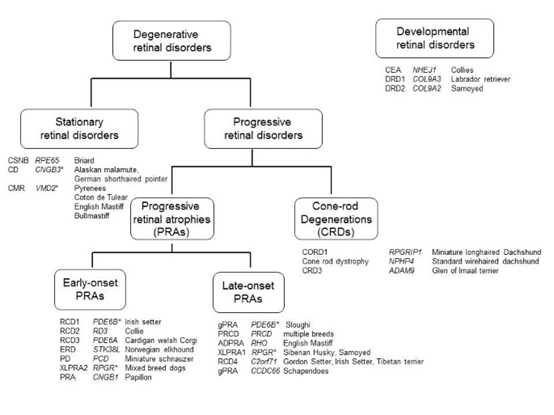
视网膜疾病
遗传形式的视网膜疾病是狗临床和遗传特征遗传条件之一。视网膜疾病可以通过多种方式进行分类。然而,大多数分类方法将广泛地考虑疾病的典型发展阶段或发病年龄,通常受影响的细胞以及疾病在狗的一生中是否逐渐变得严重,或者它是否或多或少静止。在这里,视网膜疾病大致分为两大类;视网膜正常发育然后在狗的一生中退化的退行性疾病,以及视网膜发育异常的发育或发育不良疾病。然而,应该强调的是,在某些情况下,将所有视网膜疾病分为这两种广泛的形式是广义的,少数视网膜疾病具有发育不良和退行性特征。
(图片来源于网络,侵删)
退行性视网膜疾病
大多数视网膜疾病都是退行性疾病。一些退行性疾病的特征是随着时间的推移,严重程度不可避免地增加,总是最终导致视力完全丧失,而其他疾病的特征是病理在整个生命中不会显着恶化。这两种广泛的临床疾病类别分别为进行性和平稳性。
进行性视网膜疾病
进行性视网膜萎缩(PRA)和视杆营养不良(CRD)是影响视网膜光感受器细胞的两种广泛形式的进行性双侧退行性疾病的统称。
进行性视网膜萎缩
一般来说,PRAs的特征是最初失去杆状光感受器功能,然后是视锥细胞,因此,夜盲症是大多数受PRA影响的重要临床体征。高光下的视力障碍总是随之而来,伴有眼底的特征性变化,在眼底镜检查时可见。典型的变化包括视网膜血管衰减,视网膜变薄和视盘萎缩导致锥体层反射率增加。在许多狗狗病例中,继发性白内障的发展,可能是变得足够广泛以掩盖视网膜,需要使用视网膜电图(ERG)进行诊断。虽然大多数狗表现出相同的眼底异常,但这些异常发生的年龄在品种之间差异很大,遗传上不同形式的PRA可以大致分为早发型和晚发型。
早发型 PRA
该疾病的早期发病形式通常在2至6周龄之间表达,即狗的产后视网膜分化期,并且其特征在于杆和视锥体光感受器的异常发育。四种特征明确、遗传上不同的常染色体隐性、早发性视网膜变性形式是1型视杆细胞发育不良(RCD1)、2型杆锥体发育不良(RCD2)、3型杆视锥发育不良(RCD3)和早期视网膜变性(ERD)。RCD1从出生后约25天开始影响爱尔兰Setters,并在杆细胞和视锥细胞群耗尽约1年时达到高潮,这是由cGMP磷酸二酯酶(PDE6B)β亚基的基因密码子807的无意义突变引起的,该基因是光转导途径的重要组成部分.这种突变是早期导致在疾病中鉴定出异常PRA的原因。在同一基因中,密码子816后插入8碱基对(bp)会导致Sloughi中PRA的遗传差异形式,其发病年龄晚于爱尔兰Setter形式,直到狗在2至3岁之间才注意到视力障碍的最初迹象。威尔士柯基犬的PRA,称为杆锥发育不良3(RCD3),也是由cGMP磷酸二酯酶亚基(这次是α亚基)突变引起的,该亚基导致发病年龄与RCD1相当。在RCD3受影响的狗中,正常杆介导的ERG反应无法发展,光感受器外段未达到成熟,杆细胞因细胞凋亡而丢失。遗传上不同的RCD2在粗糙光滑的牧羊犬中分离,并且是由RD3中的插入引起的,导致氨基酸的改变和扩展。RD3突变与人类和小鼠的视网膜变性有关。
虽然上述PRA,RCD1和RCD3的早发形式是在分子水平上表征的早期犬类遗传性疾病之一,但导致类似早发性疾病ERD(早发性变性)的突变直到最近才被发现。这种情况最初在挪威猎犬中描述,并在10多年前开始被绘制,是由基因STK38L中的外显子SINE插入引起的。虽然已知STK38L具有神经元细胞功能,但与异常的光感受器功能无关;与狗的这种疾病有关,使该基因成为其他物种(包括人类)中类似疾病的潜在研究对象。
一种不同形式的早发型PRA会影响微型雪纳瑞犬。从组织学上讲,这种疾病从很小的时候就很明显,当正常的视网膜接近产后分化结束时,并且由于它同时影响视杆细胞和视锥细胞,因此被称为光感受器异型增生(PD)。这种疾病早期与磷屑素(PDC)的错义突变有关。然而,此后的其他研究导致完全排除磷屑素,并鉴定出实际上导致这种疾病的基因和突变,也称为A型PRA。有证据表明,A型PRA实际上是微型雪纳瑞中一种罕见的PRA形式,其他遗传上不同的PRA形式在该品种内分离,其突变尚未确定。
最近,在具有早发PRA的CNB1的外显子26中发现了一个复杂的突变,包括一个碱基对缺失和6个碱基对插入的组合。突变导致移码和过早停止密码子。受影响的狗表现出早期缺乏杆状功能,随后出现缓慢的视网膜变性,其表型可与具有CNGB1突变的小鼠和人类相当。CNGB1与CNGA1结合形成杆环核苷酸门控通道。先前的研究表明 , CNGB1 需要 CNGA1 将 CNGA1 正常靶向到杆外段 ,并且作者确实能够证明在受影响的状蛋白纯合子的杆外节中缺乏可检测的 CNGA1 蛋白。Ahonen和同事在Phalene犬中也描述了相同的突变。
上述PRA的早期发病形式都是由常染色体基因突变引起的。相反,X连锁视网膜色素性GTP酶调节基因(RPGR)的突变导致一种非常严重的PRA形式,称为XLPRA2,该形式已在混种犬中描述。XLPRA2突变是2核苷酸缺失,导致帧移位,通过导致许多酸性谷氨酸残基被碱性精氨酸残基替换来显着改变预测的肽序列,并导致蛋白质71氨基酸的过早终止下游。与下面描述的遗传上独特的,相对晚发的XLPRA1不同,与XLPRA2中的移码突变相关的表型非常严重,并且在视网膜发育过程中表现出来。ERG异常在5-6周龄时明显,细胞变性在4个月时出现,表明突变蛋白具有毒性功能增益,严重损害光感受器发育的早期阶段。
PRA 的晚发型
PRA的晚发形式是已完成正常发育的光感受器的退化。与早发性疾病相关的基因是光感受器正常发育所必需的,而与晚发性疾病相关的基因则是这些细胞的长期维持和功能所必需的。
进行性杆锥变性(PRCD)是PRA的一种晚发形式,影响多个品种。在分子水平上对这种疾病进行表征之前,进行杂交,以确定在多个品种中分离的表型相似的疾病,包括微型贵宾犬,英国和美国可卡犬,拉布拉多猎犬,澳大利亚牧牛犬,新斯科舍省寻鸭猎犬和葡萄牙水犬。然而,当受PRCD影响的狗与受PRA影响的边境牧羊犬、巴森吉犬和意大利灰狗交配时,后代是正常的,这表明这些品种受到遗传上不同形式的疾病的影响。在犬类基因组序列可用之前,1998年将PRCD位点映射到CFA9上的一个大区域,而研究犬类基因组的工具相对不复杂。然而,在如此多的品种中分离出的遗传相同的疾病被证明是无价的,因为它允许在受影响的品种之间使用连锁不平衡映射来缩小PRCD相关区域。这导致最终在先前未知基因的第二个密码子中鉴定出单个核苷酸取代,该基因现在已知是至少18个不同品种中PRCD的原因。有趣的是,在一名患有隐性视网膜色素变性(相当于PRA的人类)的人类患者中鉴定出相同的纯合突变,并建立了新的视网膜基因PRCD,作为维持跨物种的杆状光感受器结构和功能的重要基因。
在英国獒犬中已经描述了一种遗传上独特的晚发性PRA。迄今为止,这种疾病在犬类遗传性视网膜病变中是少见的,因为它是作为常染色体显性疾病遗传的,并且是由视紫红质(RHO)核苷酸11处的单一非同义C→G转变引起的,该变性将Thr-4转变为Arg(T4R)。携带RHO突变的狗在3至6个月大时具有正常的光感受器特异性ERG功能,但到13个月时,这些反应是异常的。在年轻的受影响狗视网膜结构中,视紫红质表达和光感受器激活是正常的;疾病进展的特征是初始局灶性感光感受器变性区域被结构正常的视网膜区域包围,有趣的是,这与RHO突变的人类的表型非常相似。与 T4R 突变相关的表型的一个特征性组成部分是光照暴露与受影响动物中发生的视网膜组织早期改变之间的剂量-反应关系。最高剂量的光会导致神经元的快速丧失,在<4周内达到光感受器的完全变性,而最低剂量的光照能够使机制在数周至数月的时间尺度内起作用,以修复由神经元应激引起的异常改变。这种突变最初在英国獒犬中被发现,在PRA影响的公牛獒犬中也有发现,但迄今为止尚未在任何其他品种中被发现。
在 Schapendoes 中描述了一种不同的晚发型常染色体隐性遗传性全身性 PRA,其发病年龄通常在 2-5 岁之间。在疾病的早期阶段,受影响的狗变得夜盲,缺乏调整视力以适应昏暗光线的能力;后来在白天视力也逐步下降。这种完全性光感受器变性的过程需要长达2年的时间。该疾病的因果突变已被证明是最近发现的含有66(CCDC66)的基因盘绕线圈结构域的外显子6中的单个bp插入,导致终止密码子。CCDC66在进化上在不同的脊椎动物物种中是保守的,并且表现出复杂的差异RNA剪接模式,导致视网膜中的各种亚型。在免疫组织化学上,CCDC66蛋白主要在小鼠、狗和人类的光感受器内段检测到,尽管受影响的沙潘多斯的视网膜已被证明缺乏CCDC66蛋白。
RPGR中的突变与XLPRA2相关的突变(如上所述)不同,负责一种与性别相关的晚发型PRA,该形式最初在西伯利亚哈士奇中被描述为XLPRA1。该突变也在萨摩耶犬中被发现,是五核苷酸缺失,导致移码和立即过早停止;截断的蛋白质缺乏230C末端氨基酸,导致等电点略有下降。携带这种突变的狗的光感受器正常发育,与具有XLPRA2的狗相反,并且在形态和功能上保持正常,直到成年,这表明RPGR蛋白的C端对于杆细胞和视锥细胞的功能和结构分化并不重要。
最近在C2orf71中发现了一种移码突变,该突变导致戈登和爱尔兰塞特斯的常染色体隐性遗传性晚发PRA。所研究的狗的平均发病年龄约为10岁。该变异在21例PRA病例中有19例是纯合子的,在Gordon Setter人群中的频率约为0.37。本研究中大约10%的病例(2/21)与C2orf71突变无关,表明该品种中的PRA在遗传上是异质性的,并且由至少两个突变引起。该变体也存在于许多具有PRA的爱尔兰Setter犬中,并且该品种的等位基因频率估计为0.26。C2orf71的功能尚不清楚,但对视网膜发育和功能很重要,并且先前与人类常染色体隐性遗传性视网膜色素变有关。与C2orf71突变相关的PRA形式被称为RCD4,用于杆锥变性4,以区别于其他形式的杆锥变性。在受PRA影响的西藏梗犬中也发现了这种突变(Mellersh和Downs,未发表)。
所有被描述为晚期视网膜疾病的进行性,或多或少地表现为由高度渗透性突变引起的单基因疾病。然而,有证据表明,环境修饰剂可能在其中一些疾病中发挥作用,导致品种之间和品种内部的表型变异。
锥杆退化
视锥杆营养不良主要是视锥细胞的疾病,视杆细胞在以后会受到影响。CRD的眼底变化与PRA非常相似,需要详细的ERG研究来测量锥体和视杆特异性反应,以区分这两种类型的疾病。出于这个原因,一些疾病最初被描述为PRA,后来在进行广泛的ERG调查时被重新分类。
其中一种疾病是一种视网膜变性,在微型长毛腊肠犬(MLHD)中已有描述。该病最初被描述为一种早发性常染色体隐性PRA,所有腊肠犬均在近交系研究菌群内显示眼科异常,ERG在6周龄时可检测到,25周时通过眼底镜检查可检测到,并在2岁时失明。随后的一项视网膜电图研究发现,视锥细胞光感受器功能最初降低,导致该病症被重新分类为视锥杆营养不良(CRD),而不是视杆导向的PRA,该疾病被称为CORD1,用于锥杆变性1。同样的病症也被其他人称为CRD4,用于锥杆退化4。Lheriteau及其同事后来的发现也与CRD的病症一致。使用相同的狗群将CORD1映射到CFA15上的一个大区域,并在研究菌落中发现了RPGRIP1的突变,该突变与CORD1完全共隔离。该突变是A29束的44 bp插入,两侧是基因外显子2中的15 bp重复,这会产生帧移位并在外显子3的早期引入过早的终止密码子。RPGRIP1的突变与Leber先天性黑朦病(LCA),人类视网膜色素变性(RP)和CRD以及小鼠的遗传性视网膜异常有关,这表明它在视觉功能中起重要作用。该基因产物的确切作用目前尚不清楚,但人们认为它将调节复合物锚定在连接纤毛的光感受器上,纤毛充当光感受器细胞内段和外段之间的桥梁,并且在椎间盘形态发生和睫状轴突的结构中具有功能。RPGRIP1还与NPHP4相互作用,NPHP4是一种基因,与标准线毛腊肠犬品种中早发性CRD分离的遗传差异形式有关。在MLHD的研究群中,RPGRIP1基因型与狗的表型与其CORD1表型完全相关,而在宠物MLHD群体中则并非如此。在殖民地之外,对于RPGRIP1插入(称为RPGRIP1突变体)纯合子的狗,视网膜变性的发病年龄存在相当大的差异,这也在其他品种中被鉴定出来,包括英国斯普林格猎犬(ESS)和比格犬。在一项关于少量RPGRIP1突变小猎犬的研究中,ERG锥体反应是无法检测到的,而杆状反应在狗之间以及同一只狗的眼睛之间是可变的。在同一项研究中,即使没有眼底异常,所有RPGRIP1突变的MLHD均显示锥体反应降低,Busse及其同事也证实了这一发现。这些发现共同表明,还涉及其他突变,这些突变改变了与RPGRIP1突变相关的眼底异常的发病年龄。由于使用的原始研究菌落是由非常少量的狗开发的,因此该菌落确实有可能被固定在这些额外的位点上,因此在调查更多杂交的宠物种群之前未被发现。上述NPHP4突变导致标准线毛腊腊犬的早发性锥杆营养不良,但在Miyadera的腊肠犬研究中不存在,因此可以排除该突变。最近一项使用具有早发或晚发性脊髓1的RPGRIP1突变MLHDs的关联研究确实揭示了与早发性疾病分离的第二个位点,表明MLHD中的早发性CRD更有可能是一种双基因疾病,并且仅插入RPGRIP1就会导致晚发性CRD,尽管ERG异常可以在生命早期检测到。
在分子水平上表征的另一种形式的犬杆营养不良是crd3,用于锥杆营养不良3,在伊马尔梗犬的格伦中分离。这种疾病在3岁的受影响狗的眼底镜下变得明显,并在几年内进展为终末期视网膜变性。最近,两个研究小组几乎同时发现了这种因果突变,作为ADAM9(分解蛋白和金属蛋白酶结构域,家族成员9)的大型基因组缺失,它去除了ADAM9转录本的外显子15和16,并产生一个过早的终止密码子,预计会导致缺乏关键结构域的截断蛋白质。这一发现确立了CRD3作为类似人类疾病CORD9的真正正交物和潜在有用模型,其中发现了四种不同的ADAM9突变。
固定性视网膜疾病
上述PRA和CRD的形式都是遗传性视网膜病变,其特征在于随着时间的推移严重程度增加和视觉功能下降。在狗的一生中,进行性视网膜变化会导致完全失明。
Narfstrom及其同事在瑞典Briard中将特征良好的非进行性视网膜病变描述为静止性和先天性,导致其被称为先天性静止性夜盲症(CSNB)。自首次报告以来,该疾病也被描述为具有进行性成分,导致其也被称为遗传性视网膜营养不良。然而,CSNB和遗传性视网膜营养不良均被证明是由RPE65基因外显子5中的四核苷酸缺失引起的,这表明它们是遗传上相同的条件。RPE65参与全反式类视黄醇向11-顺式类视黄醇的转化,在没有RPE65的情况下,视觉周期被中断,导致视觉色素缺乏。这种犬病具有非常典型的临床表型;受影响的狗在至少5-6周龄时有严重的视力障碍,但至少在生命的前3-4年内保持眼底正常。年长的狗可能表现出轻微的视网膜异常,表明缓慢进行性视网膜退行性过程。视锥细胞和视杆介导的ERG反应均高度异常,可能是由于视杆细胞和视锥细胞的反应共同作用,灵敏度非常低。在CSNB受影响的狗中观察到的具有健康杆状光感受器的狗中视觉功能的独特缺乏导致了视网膜基因治疗领域的里程碑式研究。视网膜下注射表达RPE65的腺相关病毒载体可恢复视杆感光体功能并改善视觉功能,首先见于狗,随后见于人类。
视锥变性 (CD) 也不同于其他进行性疾病,因为早发性锥体变性发生在没有随后的视杆变性(这是视锥杆营养不良的特征)的情况下。在最初在阿拉斯加Malamutes中描述的cd中,受影响的幼犬在8至12周龄之间出现日盲和畏光,当狗的视网膜发育正常时,尽管这些临床症状只发生在明亮的光线下,狗在整个一生中保持眼底正常。锥体功能在6-12周龄时开始恶化,在成年犬中未被发现。然而,杆状光感受器在动物的整个生命中在功能和结构上保持正常。在CD受影响的阿拉斯加马拉穆特衍生的狗中发现了一种大的基因组缺失,该基因可去除CNGB3的所有外显子,该基因编码锥形环状核苷酸门控阳离子通道的β亚基,但有证据表明该病症在该品种中可能是遗传异质性的,因为一些狗已被确定为缺乏CNGB3缺失的日盲临床体征.在德国短毛指针中检测到同一基因的错义突变,该基因受临床上相同的等位基因影响。这些发现确立了CD是人色盲的直系物,这种疾病也称为杆单色性或全先天性色盲,其与CD有许多共同的临床特征,并且也与CNGB3突变相关。最近,通过基因替代疗法在两种犬类无色盲模型中成功恢复了视锥功能和相关显像视觉,证明了这些直系动物的潜力。
另一种通常非进展性的遗传性视网膜疾病是犬多灶性视网膜病变(CMR),这种疾病已在多个品种中得到认可,尤其是大比利牛斯山脉、Coton de Tulear、英国獒犬和牛獒犬。在 4 个月左右龄之前,眼底变化通常很明显,其特征在于视网膜抬高的多灶性区域,其中含有视网膜下浆液积聚。视网膜抬高可保持静止数年,而多灶性外视网膜萎缩常见于老年动物。Bestrophin基因(BEST1(别名VMD2))中的几种不同变体已被确定为狗中CMR的可能因果突变。在大比利牛斯山脉,英国獒犬和牛獒犬中,外显子2中的C73T突变导致过早翻译终止,将开放阅读框架限制为25个密码子,而野生型mRNA(cmr1)和Coton de Tulears中的580个密码子G482A转变将进化保守的甘氨酸残基改变为天冬氨酸(cmr2)。在拉波尼亚牧民中,在CMR受影响的狗中描述了两种编码变化;核苷酸位置1,388(c1388del)缺失,核苷酸位置1,466处取代。c1388del导致帧偏移(Pro463fs),在氨基酸490处引入新的停止密码子,而G1466T取代本身导致氨基酸序列(Gly489Val)的保守变化,预计这将改变蛋白质功能,仅具有边际意义。然而,与C1388del结合使用时,G1466T取代导致在移位读取框架(Gly489X)内氨基酸位置489处产生额外的停止密码子。由于这些突变仅在完全连锁不平衡中被发现,作者得出结论,这些变化的组合称之为cmr3的疾病。
这些突变确立了CMR作为人类黄斑营养不良症(BMD)的新型动物模型,这是一种常染色体显性遗传的儿童视网膜疾病,也是由Bestrophin基因突变引起的。
发育性疾病
视网膜发育不良是用于表示发育中的视网膜的无序增殖和不完全分化的术语,可以细分为局灶性,多灶性,地理型和总型。局灶性和多焦点类型表现为内(感觉)视网膜层组织中的线性褶皱和“玫瑰花”,而在地理形式中,视网膜发育缺陷区域较大,表现为视网膜中央混合性高反射率或低反射率的不规则或马蹄形区域。视网膜发育不良的总或广泛形式被描述为几个品种的遗传性状,包括贝德灵顿梗,Sealyham梗,拉布拉多猎犬和约克夏梗,并且与异常神经视网膜从视网膜色素上皮完全脱离导致受影响眼睛失明有关。所有形式的视网膜发育不良都是先天性的和非进行性的。视网膜发育不良似乎是作为常染色体性状遗传的,至少在那些已经研究过足够数量的个体以可靠地估计遗传模式的品种中是这样。迄今为止,尚未在任何品种的分子水平上表征分离性或非综合征形式的视网膜发育不良的遗传学,并且没有与这种情况相关的突变。
拉布拉多猎犬和萨摩耶犬中已有综合征性视网膜发育不良的报道。纯合子患犬有短肢侏儒症和一系列以视网膜完全脱离和白内障为特征的眼部改变,而杂合子犬只有局灶性或多灶性视网膜病变。育种研究确定,这两种疾病是非等位基因,它们分别被称为DRD1(1型视网膜发育不良的侏儒症,拉布拉多猎犬)和DRD2(萨摩耶犬)(这些病症以前也被称为OSD1和OSD2用于眼骨骼发育不良)。突变最近与这两种疾病有关;COL9A3 外显子 1 中的 1 碱基对插入突变与 DRD1 相关,而 COL9A2 5' 末端的 1,267 bp 缺失与 DRD2 共隔离。这两种突变都会影响其各自基因的COL3结构域,其在受累视网膜中的表达均降低。
视网膜的另一个复杂的先天性缺陷是牧羊眼异常(CEA),尽管视网膜受累是继发于与该疾病相关的原发性眼部缺陷。该疾病的主要表型因素是脉络膜的区域发育不良,脉络膜是视网膜下方的高度血管层。相关的视网膜病变(称为结肠瘤)通常可通过眼底镜检查检测到,在少数病例中,曲折的视网膜血管和多个视网膜褶皱也是如此。CEA在几个具有牧羊犬血统的放牧品种中分离,被映射到CFA37的大面积区域,其中包括40多个基因;随后,该疾病在多个密切相关的品种中分离的事实被用来减小关键疾病相关区域的大小,并将致病突变精确定位为NHEJ1基因中7.8 kb的内含子缺失,该基因跨越高度保守的结合域,几个发育中的重要基因与之结合。然而,删除导致CEA的确切机制迄今尚未确定。
遗传性白内障
晶状体是眼睛前段的透明,双凸,无血管结构,部分负责光线的折射聚焦在视网膜上。晶状体由细胞核,皮质和囊组成,并由许多致密的带状韧带悬挂,这些韧带附着在囊上并连接睫状体和晶状体赤道之间。透明度是镜片的关键特性,部分原因是晶状体纤维内没有光散射细胞器。新的晶状体纤维是从晶状体上皮的赤道细胞产生的,它们会伸长,合成结晶,最后在成为成熟的晶状体纤维时失去其细胞核。晶体占晶状体中蛋白质的90%以上,专门用于通过形成可溶的高分子量聚集体来维持透明度,这些聚集体需要在个体的一生中保持在溶液中。
白内障被简单地定义为晶状体的不透明,有多种发病原因,包括高龄和其他疾病的继发性影响,如糖尿病或进行性视网膜萎缩和创伤。原发性或遗传性白内障(HC)在狗中很常见,是失明的主要原因。据报道,多达97种不同品种的HC,据报道,与混种犬相比,约有60个品种的风险增加。不同品种中报告的遗传性白内障在晶状体内的解剖位置,发病年龄以及进行性或静止性方面各不相同,尽管在品种内白内障通常显示出显着的品种特异性。尽管受HC影响的品种数量众多,但迄今为止,只有一个基因,即转录因子HSF4,与狗的白内障发展有关。HSF4属于热休克转录因子家族,可调节热休克蛋白的表达以响应不同的应激,例如氧化剂,重金属,高温以及细菌和病毒感染。据报道,HSF4中的不同突变可引起人类常染色体显性遗传性和隐性白内障,小鼠的研究表明,在晶状体发育过程中,HSF4是正常纤维细胞分化所必需的。基因的破坏导致白内障通过多种途径发展,包括不同结晶蛋白翻译后修饰的下调或丧失。在该基因的外显子10(CFA5 g.85286582_85286583insC)中插入单个隐性核苷酸,导致移码并过早停止密码子,是斯塔福德郡斗牛梗中HC早发、双侧对称和进行性形式的原因。这种白内障从几个月龄开始发展,如果不及时治疗,总是在2-3年内进展为全白内障。该突变由波士顿梗犬共享,其中它引起临床上相同的早发性遗传性白内障(EHC),这是已知影响该品种的两种遗传上不同的白内障形式之一。该品种中与临床上变异性更强的迟发性遗传性白内障(LHC)相关的突变尚未被发现。在少数临床上具有相同白内障的法国斗牛犬中也发现了相同的突变(Mellersh,未发表)。
在HSF4(CFA5 g.85286582delC)中,相同位置的单核苷酸缺失也与澳大利亚牧羊犬中的HC有关。在斯塔福德郡斗牛梗和相关品种中发现的由插入引起的白内障形式具有隐性和高度渗透性的遗传模式,是早发,高度进展和均匀的。相比之下,在澳大利亚牧羊犬中观察到的白内障形式,由上述缺失引起,具有显性或共同显性遗传模式,并非完全渗透,通常与后极性囊下白内障有关,该白内障也具有可变的发病年龄。与白内障发展相关的其他突变很可能在澳大利亚牧羊犬种群中共同隔离,因为并非所有患有双侧后极性囊下白内障的狗都携带HSF4缺失的副本。
HSF4被排除在一长串品种的HC开发之外,包括阿拉斯加马拉穆特,美国可卡犬,比雄哈瓦那犬,比利时牧羊犬Tervueren和Groenendael,腊肠犬,英国可卡犬,英国微型梗犬,芬兰Lapphund,金毛猎犬,格里芬布鲁塞尔犬,Kromfohrlander,Jack Russell梗犬,拉蓬牧羊犬,微型雪纳瑞犬,微型Pinscher,新斯科舍省寻鸭猎犬, 罗威纳犬、萨摩耶犬、雪纳瑞犬和藏獒犬。与狗的遗传性视网膜变性相关的突变相比,文献中报道的犬类白内障突变的缺乏证明了这样一个事实: HC可能是大多数品种狗中的遗传复杂性疾病,迄今为止的研究尚未包括对足够数量的病例和对照的分析,以确定与该疾病相关的DNA变异。对于微型雪纳瑞犬中的先天性白内障和小眼炎以及恩特勒布赫山地犬,Bichon Frise和美国可卡犬中的白内障,已经提出了隐性遗传模式。相反,对于在挪威布洪德观察到的粉状(尘埃样)白内障,需要采用具有高度外显率的常染色体显性遗传模式,并且对于拉布拉多犬和金毛猎犬的遗传性后极性囊下白内障,建议使用具有可变外显率的常染色体显性遗传模式,尽管目前的证据表明,拉布拉多的白内障是作为常染色体隐性遗传性状遗传。少数其他品种的遗传证据已经报道,包括Leonberger,Jack Russell Terrier和Chow chow,尽管很少确定确切的遗传模式。
主镜头光度
初级晶状体脱位(PLL)不是晶状体本身的疾病,而是晶状体悬吊装置(区域)的遗传性恶化,该膜带是将晶状体从睫状体悬浮的纤维系统,将其保持在视觉轴内并与玻璃体的前表面接触。在受PLL超微结构异常影响的狗中,区状纤维的超微结构异常在20个月大时已经很明显,远早于通常在狗3至8岁时发生的晶状体脱位,这是由于斑带的退化和分解导致晶状体从眼内的正常位置移位.在大多数情况下,脱臼的晶状体会进入前房,其存在可能导致急性青光眼。100多年来,这种情况一直被认为是犬科疾病,并且在几个梗犬品种和其他一些可能具有共同祖先的犬种中频繁出现。PLL在西藏梗犬中是隐性遗传的,并且被认为在沙培和其他西方梗犬品种中遗传是隐性的。ADAMTS17的突变被描述为三个品种PLL的原因,即微型公牛梗,兰开夏郡高跟犬和杰克罗素梗犬。该突变是c.1473 + 1处的G→A取代,它破坏了内含子10中的剪接供体识别位点,并导致外显子跳跃,导致移码和过早终止密码子的引入。绝大多数受PLL影响的狗是突变的纯合子,少数是杂合子,人们猜测与野生型等位基因纯合的狗相比,至少某些品种的携带者患该病的风险可能更高。ADAMTS17是ADAMTS基因家族的29个已知哺乳动物成员之一,这些基因编码分泌的金属蛋白酶,这些金属蛋白酶在蛋白水解上修饰细胞外结构蛋白。多种ADAMTS基因的突变与多种人类疾病有关,包括Ehlers-Danlos综合征和Weill-Marchesani综合征。犬类ADAMTS17剪接位点突变由至少17个不同的品种共享,其中许多是梗犬或梗犬型品种,但其中一些具有更多样化的起源。一些已知患PLL风险增加的品种,如边境牧羊犬,并不携带与梗犬品种相同的ADAMTS17突变,这表明它们的疾病形式必须具有遗传差异,尽管临床上相似。
其他条件
上面描述的晶状体和视网膜疾病代表已经确定为因果突变的狗的绝大多数遗传性眼部疾病。据报道,许多其他眼部疾病在某些品种中比其他品种更常见,这表明它们具有遗传成分。然而,对于相对较少的这些条件,已经对继承方式进行了严格的估计。所描述的其余疾病仅限于那些已经报告了遗传方式或遗传性的估计条件。
青光眼
青光眼是用于描述一组导致眼内压升高的疾病,对视网膜神经节细胞及其轴突造成损害,导致视力丧失和失明。青光眼通常分为先天性,原发性和继发性类型,具体取决于病因。先天性青光眼在狗中很少见,继发性青光眼是狗中观察到的最常见形式的疾病,由于先前或并发的眼部疾病而产生,因此本身不是遗传的,尽管原发性,因果关系可能是。原发性青光眼发生在没有任何其他眼部疾病的情况下,因此,推测在大多数品种中具有遗传成分。原发性青光眼可能发生在存在(闭角型青光眼)或不存在(开角型青光眼)进入睫状裂隙的异常、狭窄或闭合的开口的情况下,这阻止了体液水从眼睛的前房通过虹膜角膜角膜通过果胶纤维之间的开口有效引流。性腺病发生是狗原发性青光眼的最常见原因,指的是存在异常,形状不规则或穿孔的果胶纤维片。据报道,青光眼在几个品种中比平均水平更为普遍,包括平衣猎犬,美国可卡犬,巴塞特猎犬,沙贝犬,挪威麋鹿和波士顿梗犬。据报告,大丹犬的性腺病发生与青光眼之间存在强烈且显著的相关性,同一项研究还报告了性腺病发生的高遗传性,表明青光眼在该品种中可能是可遗传的。据报道,扁衣猎犬的果胶韧带发育不良与成人发病的原发性青光眼之间存在类似显著的关联,其遗传力估计约为0.7。迄今为止,尚未发现与任何品种的狗的闭角型青光眼相关的突变,尽管最近在Dandie Dinmont Terriers中发现了第一个青光眼相关位点。
常染色体隐性遗传原发性开角型青光眼(POAG)在Beagle中得到了很好的表征,ADAMTS10中的Gly661Arg变体与小猎犬的病情有关,该疾病在8至16月龄时出现眼内压升高,原因是尽管正常出现开放的虹膜角角,但对房水流出的抵抗力增加。
持续性增生性原发性玻璃体
持续性原发性玻璃体增生性 (PHPV) 是一种先天性、非进行性疾病,由胎儿舌骨脉管系统异常消退引起。这种情况很少见,但在斯塔福德郡斗牛梗中常见,其中系谱分析支持该病的遗传病因,但不足以确定确切的遗传模式,。PHPV和持续性增生性静脉血管(PHTVL)也在杜宾犬中进行了详细描述。
原作者:凯瑟琳·梅勒什